




赛题名称:Open Problems – Single-Cell Perturbations 赛题类型:预测小分子如何改变不同细胞类型中的基因表达 赛题链接👇:
https://kaggle.com/competitions/open-problems-single-cell-perturbations/
比赛介绍
人类生物学可以非常复杂,部分原因在于人体约有37万亿个细胞,这些细胞组织成组织、器官和系统。然而,最近单细胞技术的进步为我们提供了对细胞和组织在DNA、RNA和蛋白质水平的功能提供了前所未有的洞察力。
然而,利用单细胞方法来开发药物需要建立化学干扰和对细胞状态的下游影响之间的因果关系。这些实验成本高昂且耗时,而且并非所有的细胞和组织都适用于高通量转录组筛选。如果数据科学能够帮助准确预测新细胞类型中的化学干扰,它可能会加速和扩大新药物的开发。
该竞赛的目标是预测小分子化合物如何在不同细胞类型中改变基因表达。你的工作将有助于开发预测细胞如何对小分子药物干扰作出反应的方法,这在药物发现和基础生物学方面可能具有重要应用。
评估指标
在这个竞赛中,评分是通过计算均方根误差的平均值来完成的,具体计算方法如下:
其中, 是得分行数的数量, 和 分别是第 行和第 列的实际值和预测值, 是列数。
在评估集中的每个 id
中,你需要为其对应的18,211个基因的每一个预测一个值,这些基因在剩余的列中被命名。每个 id
对应一个 cell_type
sm_name
对,你可以从 id_map.csv
文件中进行匹配识别。
id,A1BG,A1BG-AS1,...,ZZEF1
0,0.0,0.0,...,0.0
1,0.0,0.0,...,0.0
2,0.0,0.0,...,0.0
3,0.0,0.0,...,0.0
...
其中,每一行代表一个 id
,后面跟随着对应基因的预测值。
数据集描述
这个竞赛涉及到一个新型的人类外周血单核细胞(PBMCs)的单细胞干扰数据集。从整合网络基础细胞信号库(Library of Integrated Network-Based Cellular Signatures,LINCS)连接图数据集中选择了144种化合物,并在治疗后的24小时内测量了单细胞基因表达谱。该实验在三名健康人体供体中重复进行,并根据在CD34+造血干细胞中观察到的多样化转录标志物选择了这些化合物(数据未发布)。
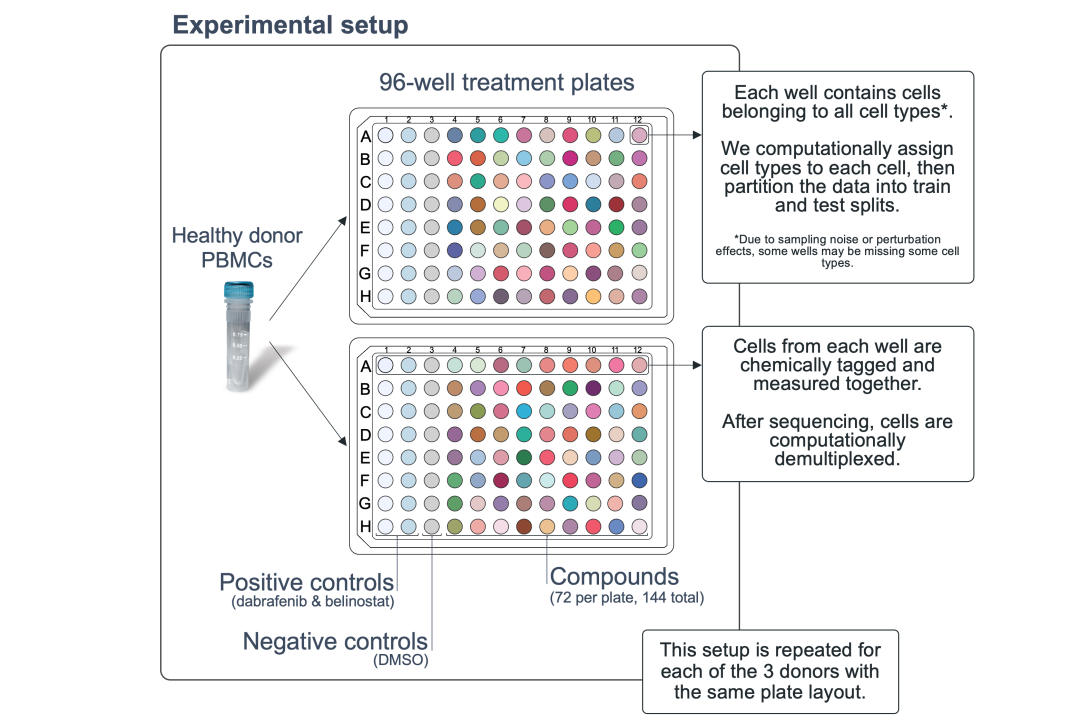
竞赛中的技术细节:
实验设计:PBMCs从供体中解冻并放置在96孔板上。板的两列用于正对照(dabrafenib和belinostat),一列用于阴性对照(DMSO)。正对照被选择,因为它们倾向于对转录产生较大影响,而阴性对照则用作该研究中使用的化合物的溶剂。板上的其余孔用于每个供体的72种化合物。整个数据集包括每个供体的2个不同的化合物板,总共有6个板。
细胞类型:每个孔中包含PBMCs,这是不同细胞类型的集合,包括T细胞、B细胞、NK细胞和髓样细胞,如巨噬细胞和单核细胞。基于单细胞RNA表达中测得的基因表达数据,我们可以计算地将每个细胞分配到一个细胞类型。需要注意的是,因为我们每孔只测量大约350个细胞,并且因为化合物可能对某些细胞类型产生毒性效应,所以我们不一定在每个孔中都观察到每种细胞类型。
化学标记:实验中的另一个技术变量是板上每行每孔的化学标记,然后将每行中的所有样本汇总到一个池中进行测序。这被称为细胞多重化,您可以在10x Genomics的网站上了解更多关于细胞多重化是什么的信息。需要了解的是,这会在板的每行中连接所有孔之间产生一些技术偏差。在板的每一行中包括两个正对照和一个阴性对照的目的之一是在计算差异表达时考虑到这种噪声源。
差异表达计算:在这个竞赛设置中,参与者的任务是建立差异表达(DE)模型,该模型可以估计实验干扰对转录中每个基因表达水平的影响(在此数据集中有18,211个基因)。我们通过首先对每个样本中每个特定类型的细胞的原始基因表达计数进行均值化来估计每个化合物的影响,这在单细胞文献中称为伪均值(pseudobulking)。然后,我们使用Limma对伪均值计数数据进行线性模型拟合,并包括库(行)、板和供体作为技术协变量,以及化合物作为实验协变量。
您的模型输入将是cell_type
和sm_name
的元组,您的模型输出将是所有18211个基因的预测signed -log10(p-values)
。
竞赛数据文件和字段描述:
de_train.parquet:稠密数组格式中的聚合差异表达数据。 基因 A1BG
、A1BG-AS1
、...、ZZEF1
(共18,211个) - 每个基因的差异表达值。cell_type
- 基于RNA表达确定的每个细胞的注释细胞类型。sm_name
- LINCS选择的(父)化合物的主要名称(标准化表示),用于将此实验中的数据映射到LINCS连接图数据。sm_lincs_id
- 全球LINCS ID(父)化合物(标准化表示
),用于将此实验中的数据映射到LINCS连接图数据。
SMILES
- 化合物的Simplified molecular-input line-entry system (SMILES)表示,这是分子结构的一维表示。这些SMILES由Cellarity根据此实验中订购的具体化合物提供。control
- 指示此实例是否用作控制的布尔值。adata_train.parquet:COO稀疏数组格式中的未聚合计数和标准化数据,是
de_train
的补充。除了de_train
中的字段外,此数据还包括:obs_id
- 为原始数据集中的每个细胞分配的唯一标识符。gene
- 对应于de_train
的列。count
- 在实验中测量的基因表达数据的原始分子计数,由10x CellRanger输出。normalized_count
- 这些计数已经进行了库大小归一化和log(X+1)变换。adata_obs_meta.csv:
adata_train
的观察元数据。library_id
- 每个库的唯一标识符,这是对来自每个板行的汇总样本的测量。同一板上同一行的所有孔中的所有细胞将共享一个library_id。plate_name
- 同一板上所有样本的唯一ID。well
- 每个板上样本的孔位置(在96孔板实验中是标准的)。这是行和列的连接。row
- 样本来自的板上的哪一行。col
- 样本来自的板上的哪一列。donor_id
- 标识样本的供体来源,共有三种。cell_type
- 基于RNA表达的每个细胞的注释细胞类型。与de_train.parquet
中的cell_type
匹配。cell_id
- 这是为了与LINCS连接图元数据保持一致而包含的,LINCS连接图元数据为每个细胞系指定一个cell_id。sm_name
- LINCS选择的(父)化合物的主要名称(标准化表示),用于将此实验中的数据映射到LINCS连接图数据。sm_lincs_id
- 全球LINCS ID(父)化合物(标准化表示),用于将此实验中的数据映射到LINCS连接图数据。SMILES
- 化合物的Simplified molecular-input line-entry system (SMILES)表示,这是分子结构的一维表示。这些SMILES由Cellarity根据此实验中订购的具体化合物提供。dose_uM
- 微摩尔尺度上的化合物剂量。对应于LINCS的pert_idose
字段。timepoint_hr
- 治疗持续时间(以小时为单位)。对应于LINCS的pert_itime
字段。control
- 指示此观察是否用作控制的布尔值。multiome_train.parquet:这是每个样本在基线时的可选额外的10x Multiome数据。
obs_id
- 每个观察的唯一标识符(与adata中使用的标识符不同)。location
- 这是一个特征ID。如果feature_type
是Gene Expression
,则这是一个基因符号。如果feature_type
是Peaks
,则这是峰值的基因组区间。count
- 这是由Cellranger-Arc输出的可访问DNA测量的原始分子计数。normalized_count
- 如果feature_type
是Gene Expression
,则这是库大小归一化和log(X+1)变换后的计数。如果feature_type
是Peaks
,则这是使用默认的log(TF) * log(IDF)
进行TF-IDF变换的ATAC-seq峰值计数。multiome_obs_meta.csv:
multiome_train.parquet
的观察元数据。obs_id
- 与multiome_train.parquet
中的相应标识符相对应。cell_type
- 基于RNA表达的每个细胞的注释细胞类型。donor_id
- 标识样本的供体来源,共有三种。multiome_var_meta.csv:
location
- 这是一个特征ID。如果feature_type
是Gene Expression
,则这是一个基因符号。如果feature_type
是Peaks
,则这是
峰值的基因组区间。
gene_id
- 这是另一种唯一的特征ID。如果feature_type
是Gene Expression
,则这是一个Ensembl Stable Gene ID。如果feature_type
是Peaks
,则这是峰值的基因组区间。feature_type
- 表示特征是RNA表达测量还是染色质可访问性测量。genome
- 运行CellRanger-Arc时使用的基因组版本。interval
- 每个特征在参考基因组上的位置。基因组坐标直接与参考基因组相关,包括染色体名称、起始位置和终止位置的格式,如chr1:1234570-1234870
。id_map.csv:标识给定
id
的cell_type
/sm_name
对。sample_submission.csv:正确格式的示例提交文件。有关更多详细信息,请参阅评估页面。
赛题赛程
2023 年 9 月 12 日 - 开始日期。 2023 年 11 月 23 日 - 报名截止日期。 2023 年 11 月 23 日 - 团队截止日期。 2023 年 11 月 30 日 - 最终截止日期。
解题思路
竞赛的目标是预测小分子化合物如何影响不同细胞类型的基因表达。为了解决这个问题,你需要使用训练数据来构建一个模型,然后使用该模型来预测测试数据中的基因表达变化。
# 竞赛交流群 邀请函 #
每天大模型、算法竞赛、干货资讯

