




点击上方↗️「活水智能」,关注 + 星标🌟

作者:David Wells
编译:活水智能
在本文中,我们将通过分析疾病本体(disease ontology)作为知识图谱,来识别和可视化不同类型的癌症聚类。具体来说,我们将在 Docker 容器中设置 Neo4j,导入本体,生成图聚类和嵌入(em)beddings),然后使用降维技术绘制这些聚类并获取一些洞见。尽管我们以 disease_ontology
作为示例,但相同的方法也可以用于探索任何本体或图数据库。
本体设置
在图数据库中,数据不是以行的形式存储(如电子表格或关系型数据库),而是存储为节点及其之间的关系。例如,在下图中,我们可以看到黑色素瘤(melanoma)和癌(carcinoma)是细胞类型癌症肿瘤(cell type cancer tumor)的子类别(通过 SCO 关系表示)。通过这种数据结构,我们可以清楚地看到黑色素瘤和癌症之间的关联,即使数据本身并未明确指出这一点。

本体(Ontology)是一组正式定义的概念及其关系。相比自由文本,本体更易于计算机解析,因此更容易从中提取有意义的信息。本体在生物科学领域被广泛使用,您可以在 OBO Foundry 上找到感兴趣的本体。在这里,我们关注的是疾病本体(Disease Ontology),它展示了不同类型疾病之间的关系。
Neo4j 是一个用于管理、查询和分析图数据库的工具。为了简化设置,我们将使用 Docker 容器。
docker run \
-it - rm \
- publish=7474:7474 - publish=7687:7687 \
- env NEO4J_AUTH=neo4j/123456789 \
- env NEO4J_PLUGINS='["graph-data-science","apoc","n10s"]' \
neo4j:5.17.0
在上述命令中,-publish
选项用于设置端口,以便 Python 直接查询数据库,同时允许我们通过浏览器访问数据库。NEO4J_PLUGINS
参数指定要安装的插件。不幸的是,Windows 版 Docker 镜像无法正确安装这些插件,因此如果您使用 Windows,则需要手动安装 Neo4j Desktop。不过,不必担心,其他步骤仍然适用于您。
当 Neo4j 运行时,您可以在浏览器中访问 http://localhost:7474/ 来连接数据库,或者使用 Python 驱动程序进行连接,如下所示。请注意,我们使用的是在 Docker 命令中发布的端口,并使用之前定义的用户名和密码进行身份验证。
URI = "bolt://localhost:7687"
AUTH = ("neo4j", "123456789")
driver = GraphDatabase.driver(URI, auth=AUTH)
driver.verify_connectivity()
在设置好 Neo4j 数据库后,我们需要导入一些数据。Neo4j 插件 n10s
旨在导入和处理本体数据;您可以使用它将数据嵌入到现有本体中,或者直接探索本体结构。以下 Cypher 命令首先设置了一些配置以优化查询结果,然后定义了唯一性约束,最后导入疾病本体数据。
CALL n10s.graphconfig.init({ handleVocabUris: "IGNORE" });
CREATE CONSTRAINT n10s_unique_uri FOR (r:Resource) REQUIRE r.uri IS UNIQUE;
CALL n10s.onto.import.fetch(http://purl.obolibrary.org/obo/doid.owl, RDF/XML);
如果您想了解如何使用 Python 驱动程序完成此操作,可以查看完整代码:GitHub 代码链接。
现在,我们已经成功导入了本体,您可以在浏览器中打开 http://localhost:7474/ 来手动探索本体结构。但我们更关注整体趋势,因此接下来我们将进行分析,具体来说,我们将执行 Louvain 聚类并生成快速随机投影(Fast Random Projection)嵌入。
聚类与嵌入
Louvain 聚类是一种用于网络数据的聚类算法。简而言之,它识别出在图中彼此连接更紧密的节点集合,并将这些节点定义为一个聚类。当应用于本体时,它可以快速识别一组相关概念。而快速随机投影(Fast Random Projection)则为每个节点生成一个嵌入,即一个数值向量,其中相似的节点具有更相似的向量。借助这些工具,我们可以识别哪些疾病相似,并量化它们的相似程度。
为了生成嵌入和聚类,我们需要“投影”出图中我们感兴趣的部分。由于本体通常非常庞大,这种子集筛选方法可以加快计算速度,并避免内存溢出。在本示例中,我们仅关注癌症,而不涉及其他类型的疾病。我们使用以下 Cypher 查询来筛选数据:匹配带有“cancer”标签的节点,以及通过 SCO 或 SCO_RESTRICTION 关系与其相关的节点。此外,我们还希望包含癌症类型之间的关系,因此添加了第二个 MATCH
查询来返回这些连接的节点及其关系。
MATCH (cancer:Class {label:"cancer"})<-[:SCO|SCO_RESTRICTION *1..]-(n:Class)
WITH n
MATCH (n)-[:SCO|SCO_RESTRICTION]->(m:Class)
WITH gds.graph.project(
"proj", n, m, {}, {undirectedRelationshipTypes: ['*']}
) AS g
RETURN g.graphName AS graph, g.nodeCount AS nodes, g.relationshipCount AS rels
一旦完成投影(我们将其命名为“proj”),我们就可以计算聚类和嵌入,并将其写回原始图数据库。最后,我们查询图数据库,获取每种癌症类型的新嵌入和聚类,并将其导出为 CSV 文件。
CALL gds.fastRP.write(
'proj',
{embeddingDimension: 128, randomSeed: 42, writeProperty: 'embedding'}
) YIELD nodePropertiesWritten
CALL gds.louvain.write(
"proj",
{writeProperty: "louvain"}
) YIELD communityCount
MATCH (cancer:Class {label:"cancer"})<-[:SCO|SCO_RESTRICTION *0..]-(n)
RETURN DISTINCT
n.label as label,
n.embedding as embedding,
n.louvain as louvain
结果
让我们查看一些聚类,看看哪些类型的癌症被归为一组。在 Python 中加载导出的数据并存入 Pandas DataFrame 后,我们可以检查单个聚类的组成情况。
聚类 2168 包含一组胰腺癌。
nodes[nodes.louvain == 2168]["label"].tolist()
#array(['"islet cell tumor"',
# '"non-functioning pancreatic endocrine tumor"',
# '"pancreatic ACTH hormone producing tumor"',
# '"pancreatic somatostatinoma"',
# '"pancreatic vasoactive intestinal peptide producing tumor"',
# '"pancreatic gastrinoma"', '"pancreatic delta cell neoplasm"',
# '"pancreatic endocrine carcinoma"',
# '"pancreatic non-functioning delta cell tumor"'], dtype=object)
聚类 174 是一个较大的癌症群体,但主要由癌(carcinoma)组成。
nodes[nodes.louvain == 174]["label"]
#array(['"head and neck cancer"', '"glottis carcinoma"',
# '"head and neck carcinoma"', '"squamous cell carcinoma"',
#...
# '"pancreatic squamous cell carcinoma"',
# '"pancreatic adenosquamous carcinoma"',
#...
# '"mixed epithelial/mesenchymal metaplastic breast carcinoma"',
# '"breast mucoepidermoid carcinoma"'], dtype=object)p
这些聚类是合理的,它们基于器官或癌症类型进行分组,并且对可视化分析非常有用。然而,嵌入仍然是高维的,无法直接进行可视化。幸运的是,TSNE 是一种非常有效的降维方法。在这里,我们使用 TSNE 将 128 维的嵌入降至 2 维,同时保持相似节点靠近。我们可以通过绘制散点图并按 Louvain 聚类着色来验证降维效果。如果两种方法结果一致,我们应该会看到颜色相近的节点聚集在一起。
from sklearn.manifold import TSNE
nodes = pd.read_csv("export.csv")
nodes['louvain'] = pd.Categorical(nodes.louvain)
embedding = nodes.embedding.apply(lambda x: ast.literal_eval(x))
embedding = embedding.tolist()
embedding = pd.DataFrame(embedding)
tsne = TSNE()
X = tsne.fit_transform(embedding)
fig, axes = plt.subplots()
axes.scatter(
X[:,0],
X[:,1],
c = cm.tab20(Normalize()(nodes['louvain'].cat.codes))
)
plt.show()
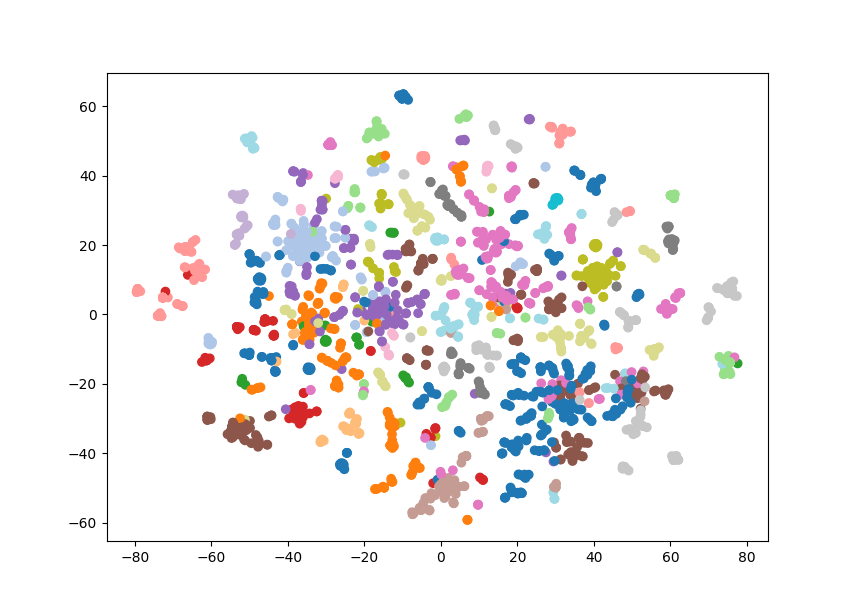
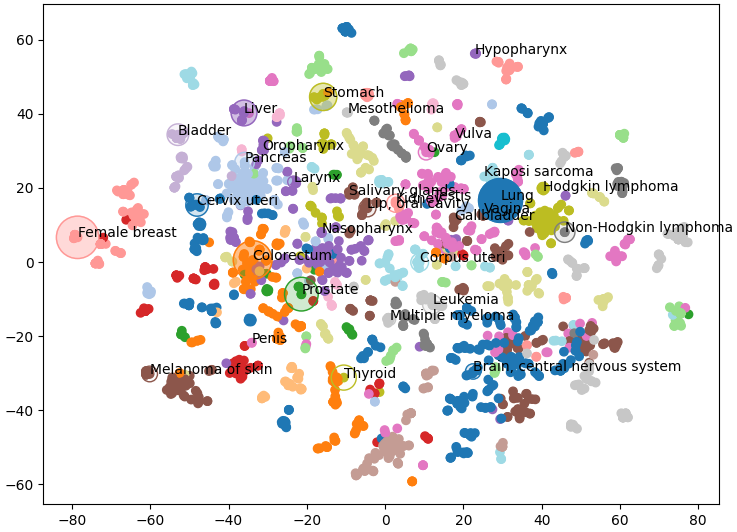
正如我们所见,相似类型的癌症聚集在一起,并且在图中以相同颜色呈现。需要注意的是,一些颜色相同的节点相距较远,这是因为我们在 29 个聚类中只能使用 20 种颜色。该可视化为我们的知识图谱提供了良好的整体视角,但我们还可以添加更多数据。
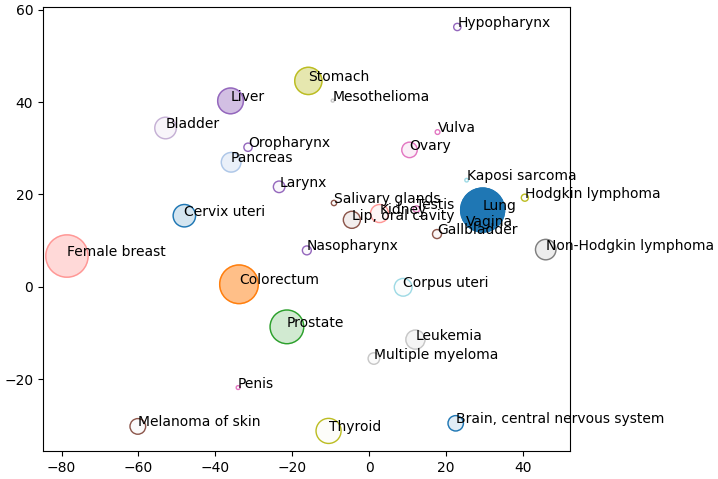
下图中,我们将癌症类型的发生频率映射为节点大小,并将死亡率映射为透明度(数据来源:Bray et al. 2024)。由于我仅获取了部分癌症类型的数据,因此仅绘制了这些节点。从图中可以看到,肝癌的整体发病率并不特别高。然而,在其所在的聚类(紫色)中,与咽喉癌、喉癌和鼻咽癌相比,肝癌的发病率明显更高。
结论
在本文中,我们利用疾病本体将不同类型的癌症分组为聚类,从而为这些疾病提供了比较的背景。希望这个小项目能帮助您了解如何可视化探索本体,并将这些信息整合到您的数据分析中。
完整代码可在 GitHub (https://github.com/DAWells/do_onto) 获取。
参考文献
Bray, F., Laversanne, M., Sung, H., Ferlay, J., Siegel, R. L., Soerjomataram, I., & Jemal, A. (2024). Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians, 74(3), 229–263.
学习资源
若要了解更多知识图谱或图数据库相关教学,你可以查看公众号的其他文章:
Neo4j+Milvus双剑合璧!打造更强大的GraphRAG知识图谱 Neo4j GraphRAG:1个Python包,轻松搞定RAG + 知识图谱! Neo4j + LangChain:如何构建基于知识图谱的最强RAG系统? 利用AI大模型,将任何文本语料转化为知识图谱,可本地运行 解读 Graph RAG:从大规模文档中发现规律,找到相互关系,速度更快,信息更全面! 利用LLM构建非结构化文本的知识图谱
活水智能成立于北京,致力于通过AI教育、AI软件和社群提高知识工作者生产力。中国AIGC产业联盟理事。
活水现有AI线下工作坊等10+门课程,15+AI软件上线,多款产品在研。知识星球中拥有2600+成员,涵盖大厂程序员、公司高管、律师等各类知识工作者。在北、上、广、深、杭、重庆等地均有线下组织。
欢迎加入我们的福利群,每周都有一手信息、优惠券发放、优秀同学心得分享,还有赠书活动~
👇🏻👇🏻👇🏻

