




通过blast、hhblits或者其他方式构建的多序列比对(MSA)都存在大量的冗余,这些冗余的序列导致多序列比对的多样性较少并相较于其他序列来说提供较少的信息,因为冗余序列的存在如果所有的序列拥有相同的权重是不合理的,通常采用序列加权方法用于减少多序列比对中的冗余序列并维持序列的多样性,这里介绍常用的Henikoff权重体系对序列进行加权。
Henikoff权重体系原理
Henikoff认为可以通过序列上每一个位点在MSA中残基的类型和数目给每个位点分配权重,通过给每个位置不同的残基类型分配相同的权重,然后在拥有相同残基的序列中平均的分配这个权重用于代表这个位置的多样性,分别给MSA中的每个位点进行权重最终实现给每条序列加权。
如下图所示,对于位点1来说,残基仅有G,出现的次数却有4次,所以权重是1/(1*4),而对于位点2来说,残基有Y, F两种,数目分别是3次和1次,首先对残基给与相同的权重1/2,然后拥有相同残基的序列平分这个权重,所以对于残基Y的权重是1/(2*3),而残基F的权重是1/(2*1),以此计算每个位点的权重,每条序列的权重即为这条序列所有位点的权重加和除以所有序列所有位点权重的加和。
对于下图的四条序列来说,可以看到对序列进行权重之后,序列3的权重最低,原因是对于每个位点来说,序列3中每个位点的残基在MSA中每列中出现次数都是较多的导致其拥有较低的权重从而导致整个序列的权重较低,说明通过这种权重方式可以很好的消除MSA中序列冗余的情况。
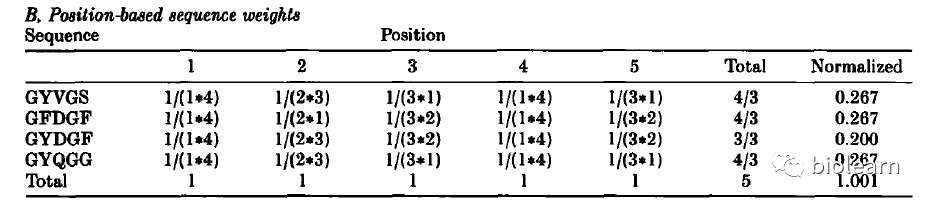
Henikoff权重体系计算方法
计算公式
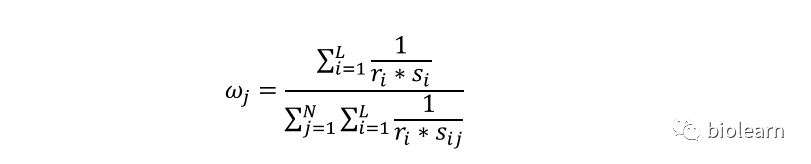
其中 N 表示MSA中序列的总数;L 表示序列的长度;i 表示序列中第 i 个序列位置;j 表示MSA中第 j 条序列;ri 表示在 i 位置上不同残基类型的数目,si 表示对应残基出现的数目,s_ij 表示第 j 条序列第 i 位置上残基在第 i 列出现的数目。
对应上图的例子,此公式中的分母的值对应的即为每条序列每个位置权重的加和,可以看到最终的值即为序列的长度,因为在MSA中每列的权重加和都为1,例如上图的Total为5,所以计算公式可以进一步简化为
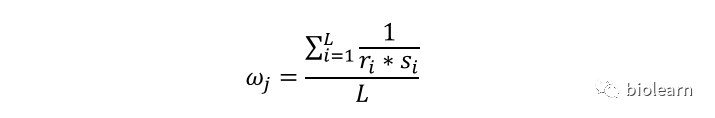
计算代码(perl脚本)
例子MSA
RPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDICGRKFARSDERKRHTKIHLR
RPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDICGRKFARSDERKRHTKIHLR
RPYACPVESCDRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDICGRKFARSDERKRHTKIHLR
RPYACPVETCVRRFSRSDELTRHIRIHTGQKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFPCDICGRKFARSDERKRHTKIHLR
RPYACPVENCDRRFSRSDELTRHIRIHTGQKPFQCRICMRSFSRSDHLTTHIRTHTGEKPFQCDTCGRKFARSDEKKRHAKVHLK
RPRKYPNRASKTPFSRSDELSRHLRIHTGHKPFQCRICMRSFSRSDHLTTHIRTHTGEKPFSCEQCGRKFARSDERRRHMRIHLR
RPYPCPAEGCDRRFSRSDELTRHIRIHTGHKPFQCRICMRNFSRSDHLTTHIRTHTGEKPFACDFCGRK----------------
KPYHCDI--CGKSFSRSDHLTTHKHIHTRERPYHCDICGKSFSVDSHLTTHKRIHAGEKPYQCDICGKSFSQTNILTTHKRIH--
KPYHCDI--CGKSFIESGQLTRHRRSHTGEKPYPCDICGRPFSYRSTFNKHRRIHTGEMP-------------------------
脚本
#!/usr/bin/perl -w
use strict;
use warnings;
open(IN,"<","F:/msa_test.txt") or die "$!";
chomp(my @msa = <IN>);
close IN;
my %aanum;
foreach my $j(@msa) {
my @tem = split(//,$j);
foreach my $i(0..$#tem) {
$aanum{$i}{$tem[$i]} ++;
}
}
foreach my $t(0..$#msa) {
my @tem = split(//,$msa[$t]);
my $numerator = 0;
foreach my $i(0..$#tem) {
my $aatype = scalar(keys%{$aanum{$i}});
my $numres = $aanum{$i}{$tem[$i]};
$numerator += 1/($aatype * $numres);
}
my $weight = $numerator/scalar@tem;
print "$t\t$weight\n";
}
运行结果
0 0.070844070961718 #第 1 条序列的权重
1 0.070844070961718 #第 2 条序列的权重
2 0.070844070961718 #第 3 条序列的权重
3 0.0762689075630252 #第 4 条序列的权重
4 0.0856676003734827 #第 5 条序列的权重
5 0.126055088702147 #第 6 条序列的权重
6 0.100401493930906 #第 7 条序列的权重
7 0.183715219421102 #第 8 条序列的权重
8 0.215359477124183 #第 9 条序列的权重
可以看到前5条非常相似的序列的权重都很低,可以有效的排除MSA中序列的冗余并提高多样性~
参考文献
Henikoff,S. and Henikoff,J.G. (1994) Position-based sequence weights. J.Mol. Biol., 243, 574–578.

 微信ID:biolearn
微信ID:biolearn
 点击下方“阅读全文”了解更多资讯
点击下方“阅读全文”了解更多资讯
