




上一篇介绍了SQL引擎源解析中“6.3.1 查询重写”、“6.3.2 统计信息和代价估算”、“6.3.3 物理路径”和“6.3.4 动态规划”的精彩内容,本章继续介绍“6.3.5 遗传算法”和“6.4 小结”的相关内容。
6.3.5 遗传算法
遗传算法(genetic algorithm,GA)作为进化算法的一种,借鉴了达尔文生物进化论中的自然选择以及遗传学机理。通过模拟大自然中“物竞天择,适者生存”这种进化过程来生成最优的个体。
当生成一定数量的原始个体后,可以通过基因的排列组合产生新的染色体,再通过染色体的杂交和变异获得下一代染色体。为了筛选出优秀的染色体,需要通过建立适应度函数计算出适应度的值,从而将适应度低的染色体淘汰。如此,通过个体间不断的遗传、突变,逐渐进化出最优秀的个体。将这个过程代入解决问题,个体即为问题的解。通过遗传算法,可以通过此类代际遗传来使得问题的解收敛于最优解。
区别于动态规划将问题分解成若干独立子问题求解的方法,遗传算法是一个选择的过程,它通过将染色体杂交构建新染色体的方法增大解空间,并在解空间中随时通过适应度函数进行筛选,推举良好基因,淘汰掉不良的基因。这就使得遗传算法获得的解不会像动态规划一样,一定是全局最优解,但可以通过改进杂交和变异的方式,来争取尽量的靠近全局最优解。
得益于在多表连接中的效率优势,在openGauss数据库中,遗传算法是动态规划方法的有益补充。只有在Enable_geqo参数打开,并且待连接的RelOptInfo的数量超过Geqo_threshold(默认12个)的情况下,才会使用遗传算法。
遗传算法的实现有下面5个步骤。
(1) 种群初始化:对基因进行编码,并通过对基因进行随机的排列组合,生成多个染色体,这些染色体构成一个新的种群。另外,在生成染色体的过程中同时计算染色体的适应度。
(2) 选择染色体:通过随机选择(实际上通过基于概率的随机数生成算法,这样能倾向于选择出优秀的染色体),选择出用于交叉和变异的染色体。
(3) 交叉操作:染色体进行交叉,产生新的染色体并加入种群。
(4) 变异操作:对染色体进行变异操作,产生新的染色体并加入种群。
(5) 适应度计算:对不良的染色体进行淘汰。
举个例子,如果用遗传算法解决货郎问题(TSP),则可以将城市作为基因,走遍各个城市的路径作为染色体,路径的总长度作为适应度,适应度函数负责筛选掉比较长的路径,保留较短的路径。算法的步骤如下。
(1) 种群初始化:对各个城市进行编号,将各个城市根据编号进行排列组合,生成多条新的路径(染色体),然后根据各城市间的距离计算整体路径长度(适应度),多条新路径构成一个种群。
(2) 选择染色体:选择两个路径进行交叉(需要注意交叉生成新染色体中不能重复出现同一个城市),对交叉操作产生的新路径计算路径长度。
(3) 变异操作:随机选择染色体进行变异(通常方法是交换城市在路径中的位置),对变异操作后的新路径计算路径长度。
(4) 适应度计算:对种群中所有路径进行基于路径长度由小到大排序,淘汰掉排名靠后的路径。
openGauss数据库的遗传算法正是模拟了解决货郎问题的方法,将RelOptInfo作为基因、最终生成的连接树作为染色体、连接树的总代价作为适应度,适应度函数则是基于路径的代价进行筛选,但是openGauss数据库的连接路径的搜索和货郎问题的路径搜索略有不同,货郎问题不存在路径不通的问题,两个城市之间是相通的,可以计算任何两个城市之间的距离,而数据库中由于连接条件的限制,可能两个表无法正常连接,或者整个连接树都无法生成。另外,需要注意的是,openGauss数据库的基因算法实现方式和通常的遗传算法略有不同,在于其没有变异的过程,只通过交叉产生新的染色体。
openGauss数据库遗传算法的总入口是geqo函数,输入参数为root(查询优化的上下文信息)、number_of_rels(要进行连接的RelOptInfo的数量)、initial_rels(所有的基表)。
1. 文件结构
遗传算法作为相对独立的优化器模块,拥有自己的一套文件目录结构,见表6-17。
表6-17 优化器文件目录结构说明文件名称 | 功能说明 |
|---|---|
geqo_copy.cpp | 复制基因函数,即gepo_copy函数 |
geqo_cx.cpp | 循环交叉(CYCLE CROSSOVER)算法函数,即cx函数 |
geqo_erx.cpp | 基于边重组交叉(EGDE RECOMBINATION CROSSOVER)实现,提供调用gimme_edge_table函数 |
geqo_eval.cpp | 主要进行适应度计算,调用make_one_rel函数生成连接关系 |
geqo_main.cpp | 遗传算法入口,即主函数geqo函数 |
geqo_misc.cpp | 遗传算法信息打印函数,辅助功能 |
geqo_mutation.cpp | 基因变异函数,在循环交叉cx函数失败时调用,即geqo_mutation函数 |
geqo_ox1.cpp | 顺序交叉(ORDER CROSSOVER)算法方式一函数,即ox1函数 |
geqo_ox2.cpp | 顺序交叉(ORDER CROSSOVER)算法方式二函数,即ox2函数 |
geqo_pmx.cpp | 部分匹配交叉(PARTIALLY MATCHED CROSSOVER)算法函数,即pmx函数 |
geqo_pool.cpp | 处理遗传算法的基因池,基因池是表示所有个体(包括染色体和多表连接后得到的新的染色体)的集合 |
geqo_px.cpp | 位置交叉(POSITION CROSSOVER)算法函数,即px函数 |
geqo_random.cpp | 遗传算法的随机算法函数,用来随机生成变异内容 |
geqo_recombination.cpp | 遗传算法初始化群体函数,即init_tour函数 |
geqo_selection.cpp | 遗传算法随机选择个体函数,即geqo_selection函数 |
这些文件作为优化器遗传算法的各个模块都在src/gausskernel/optimizer/gepo下。接下来的几个单元会着重根据这些文件中的代码进行解读。
2. 种群初始化
在使用遗传算法前,可以利用参数Gepo_threshold的数值来调整触发的条件。为了方便代码解读,将这个边界条件降低至4(即RelOptInfo数量或者说基表数量为4的时候就尝试使用遗传算法)。下面在解读代码的过程中,以t1,t2,t3,t4四个表为例进行说明。
RelOptInfo作为遗传算法的基因,首先需要进行基因编码,openGauss数据库采用实数编码的方式,也就是用{1,2,3,4}分别代表t1,t2,t3,t4这4个表。
然后通过gimme_pool_size函数来获得种群的大小,种群的大小受Geqo_pool_size和Geqo_effort两个参数的影响,种群用Pool结构体进行表示,染色体用Chromosome结构体来表示。代码如下:
/* 染色体Chromosome结构体 */
typedef struct Chromosome {
/* string实际是一个整型数组,它代表基因的一种排序方式,也就对应一棵连接树 */
/* 例如{1,2,3,4}对应的就是t1 JOIN t2 JOIN t3 JOIN t4 */
/* 例如{2,3,1,4}对应的就是t2 JOIN t3 JOIN t1 JOIN t4 */
Gene* string;
Cost worth; /* 染色体的适应度,实际上就是路径代价 */
} Chromosome;
/* 种群Pool结构体 */
typedef struct Pool {
Chromosome *data; /* 染色体数组,数组中每个元组都是一个连接树 */
int size; /* 染色体的数量,即data中连接树的数量,由gimme_pool_size生成 */
int string_length; /* 每个染色体中的基因数量,和基表的数量相同 */
} Pool;
另外通过gimme_number_generations函数来获取染色体交叉的次数,交叉的次数越多则产生的新染色体也就越多,也就更可能找到更好的解,但是交叉次数多也影响性能,用户可以通过Geqo_generations参数来调整交叉的次数。
在结构体中确定的变量如下。
(1) 通过gimme_pool_size确定的染色体的数量(Pool.size)。
(2) 每个染色体中基因的数量(Pool.string_length),和基表的数量相同。
然后就可以开始生成染色体,染色体的生成采用的是Fisher-Yates洗牌算法,最终生成Pool.size条染色体。具体的算法实现如下:
/* 初始化基因序列至{1,2,3,4} */
for (i = 0; i < num_gene; i++)
tmp[i] = (Gene)(i + 1);
remainder = num_gene - 1; /* 定义剩余基因数 */
/* 洗牌方法实现,多次随机挑选出基因,作为基因编码的一部分 */
for (i = 0; i < num_gene; i++) {
/* choose value between 0 and remainder inclusive */
next = geqo_randint(root, remainder, 0);
/* output that element of the tmp array */
tour[i] = tmp[next]; /* 基因编码 */
/* and delete it */
tmp[next] = tmp[remainder]; /* 将剩余基因序列更新 */
remainder--;
}
表6-18是生成一条染色体的流程,假设4次的随机结果为{1,1,1,0}。
表6-18 生成染色体的流程
基因备选集 | 结果集 | 随机数 | 随机数 | 说明 |
|---|---|---|---|---|
1 2 3 4 | 2 | 0~3 | 1 | 随机数为1,结果集的第一个基因为tmp[1],值为2,更新备选集tmp,将未被选中的末尾值放在前面被选中的位置上 |
1 4 3 | 2 4 | 0~2 | 1 | 随机数为1,结果集的第二个基因为4,再次更新备选集tmp |
1 3 | 2 4 3 | 0~1 | 1 | 随机数为1,结果集的第三个基因为3,由于末尾值被选,无须更新备选集 |
1 | 2 4 3 1 | 0~0 | 1 | 最后一个基因为1 |
在多次随机生成染色体后,得到一个种群,假设Pool种群中共有4条染色体,用图来描述其结构,如图6-13所示。

图6-13 染色体结构
然后对每条染色体计算适应度(worth),计算适应度的过程实际上就是根据染色体的基因编码顺序产生连接树,并对连接树求代价的过程。
在openGauss数据库中,每个染色体都默认使用的是左深树,因此每个染色体的基因编码确定后,它的连接树也就随之确定了,比如针对{2, 4, 3, 1}这样一条染色体,它对应的连接树就是(((t2,t4),t3), t1),如图6-14所示。

图6-14 染色体连接树
openGauss数据库通过geqo_eval函数来生成计算适应度,它首先根据染色体的基因编码生成一棵连接树,然后计算这棵连接树的代价。
遗传算法使用gimme_tree函数来生成连接树,函数内部递归调用了merge_clump函数。merge_clump函数将能够进行连接的表尽量连接并且生成连接子树,同时记录每个连接子树中节点的个数。再将连接子树按照节点个数从高到低的顺序记录到clumps链表。代码如下:>
/* 循环遍历所有的表,尽量将能连接的表连接起来 */
For (rel_count = 0; rel_count < num_gene; rel_count++) {
int cur_rel_index;
RelOptInfo* cur_rel = NULL;
Clump* *cur_clump = NULL;
/* tour 代表一条染色体,这里是获取染色体里的一个基因,也就是一个基表 */
cur_rel_index = (int) tour[rel_count];
cur_rel = (RelOptInfo *) list_nth(private->initial_rels, cur_rel_index - 1);
/* 给这个基表生成一个Clump,size=1代表了当前Clump中只有一个基表*/
cur_clump = (Clump*)palloc(sizeof(Clump));
cur_clump->joinrel = cur_rel;
cur_clump->size = 1;
/* 开始尝试连接,递归操作,并负责记录Clump到clumps链表 */
clumps = merge_clump(root, clumps, cur_clump, false);
}
以之前生成的{2, 4, 3, 1}这样一条染色体为例,并假定。
(1) 2和4不能连接。
(2) 4和3能够连接。
(3) 2和1能够连接。
在这些条件下,连接树生成的过程见表6-19。
表6-19 连接树生成过程
轮数 | 连接结果集 | 说明 |
|---|---|---|
初始 | NULL | 创建基因为2的节点cur_clump,cur_clump.size = 1 |
0 | {2} | 因为clumps == NULL,cur_clump没有连接表,将cur_clump直接加入clumps |
1 | {2},{4} | 创建基因为4的节点cur_clump,cur_clump.size = 1,将基因为4的cur_clump和clumps链表的里的节点尝试连接,因为2和4不能连接,节点4也被加入clumps |
2 | {2} | 创建基因为3的节点cur_clump,cur_clump.size = 1,遍历clumps链表,分别尝试和2、4进行连接,发现和4能进行连接,创建基于3和4的连接的新old_clumps节点,ols_clumps.size = 2,在clumps链表中删除节点4 |
{3, 4} {2} | 用2和4连接生成的新的old_clumps作为参数递归调用merge_clump,用old_clumps和clumps链表里的节点再尝试连接,发现不能连接(即{3,4}和{2}不能连接),那么将old_clumps加入clumps,因为old_clumps.size目前最大,插入clumps最前面 | |
3 | {3, 4} | 创建基因为1的节点cur_clump,cur_clump.size = 1 遍历clumps链表,分别尝试和{3,4}、{2}进行连接,发现和2能进行连接,创建基于1和2的新old_clumps节点,ols_clumps.size = 2,在clumps链表中删除节点2 |
{3, 4} {1, 2} | 用1和2连接生成的新的old_clumps作为参数递归调用merge_clump,用old_clumps和clumps链表里的节点尝试连接,发现不能连接,将old_clumps加入clumps,因为old_clumps.size = 2,插入clumps最后 |
结合例子中的步骤,可以看出merge_clumps函数的流程就是不断的尝试生成更大的clump。
/* 如果能够生成连接,则通过递归尝试生成节点数更多的连接 */
if (joinrel != NULL) {
…………
/* 生成新的连接节点,连接的节点数增加 */
old_clump->size += new_clump->size;
pfree_ext(new_clump);
/* 把参与了连接的节点从clumps连表里删除 */
clumps = list_delete_cell(clumps, lc, prev);
/* 以clumps和新生成的连接节点(old_clump)为参数,继续尝试生成连接 */
return merge_clump(root, clumps, old_clump, force);
}
根据上方表中的示例,最终clumps链表中有两个节点,分别是两棵连接子树,然后将force设置成true后,再次尝试连接这两个节点。
/* clumps中有多个节点,证明连接树没有生成成功 */
if (list_length(clumps) > 1) {
……
foreach(lc, clumps) {
Clump* clump = (Clump*)lfirst(lc);
/* 设置force参数为true,尝试无条件连接 */
fclumps = merge_clump(root, fclumps, clump, true);
}
clumps = fclumps;
}
3. 选择算子
在种群生成之后,就可以进行代际遗传优化,从种群中随机选择两个染色做交叉操作,这样就能产生一个新的染色体。
由于种群中的染色体已经按照适应度排序了,对来说适应度越低(代价越低)的染色体越好,因为希望将更好的染色体遗传下去,所以需要在选择父染色体和母染色体的时候更倾向于选择适应度低的染色体。在选择过程中会涉及倾向(bias)的概念,它在算子中是一个固定的值。当然,bias的值可以通过参数Geqo_selection_bias进行调整(默认值为2.0)。
/* 父染色体和母染色体通过linear_rand函数选择 */
first = linear_rand(root, pool->size, bias);
second = linear_rand(root, pool->size, bias);
要生成基于某种概率分布的随机数( x ) (x )(x),需要首先知道概率分布函数或概率密度函数,openGauss数据库采用的概率密度函数(probability density function,PDF)f x ( x ) 为:
通过概率密度函数获得累计分布函数(cumulative distribution function,CDF)F x ( x ) :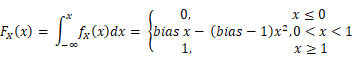
然后通过概率分布函数根据逆函数法可以获得符合概率分布的随机数。对于函数 求逆函数
求逆函数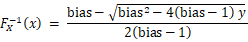
这和源代码中linear_rand函数的实现是一致的。
/* 先求的值 */
double sqrtval;
sqrtval = (bias * bias) - 4.0 * (bias - 1.0) * geqo_rand(root);
if (sqrtval > 0.0)
sqrtval = sqrt(sqrtval);
/* 然后计算的值,其为基于概率分布随机数且符合[0,1]分布 */
/* max是种群中染色体的数量 */
/* index就是满足概率分布的随机数,且随机数的值域在[0, max] */
index = max * (bias - sqrtval) / 2.0 / (bias - 1.0);
把基于概率的随机数生成算法的代码提取出来单独进行计算验证,看一下它生成随机数的特点。设bias = 2.0,然后利用概率密度函数计算各个区间的理论概率值进行分析,比如对于0.6~0.7的区间,计算其理论概率如下。
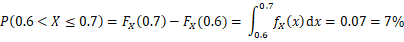
各个区间的理论概率值如图6-15所示。

图6-15 随机数生成理论概率值
从图6-15可以看出各个区间的理论概率值的数值是依次下降的,也就是说在选择父母染色体的时候是更倾向于选择适应度更低(代价更低)的染色体。
4. 交叉算子
通过选择算子选择出父母染色体之后,则可以对选出的父母染色体进行交叉操作,生成新的子代染色体。
openGauss提供了多种交叉方法,包括有基于边的重组交叉方法(edge combination crossover)、部分匹配交叉方法(partially matched crossover)、循环交叉(cycle crossover)、位置交叉(position crossover)、顺序交叉(order crossover)等。在源代码分析的过程中,以位置交叉方法为例进行说明。
假如选择父染色体的基因编码为{1, 3, 2, 4},适应度为100,母染色体的基因编码为{2, 3, 1, 4},适应度为200,在子染色体还没有生成,处于未初始化状态时,这些染色体的状态如图6-16所示。

图6-16 染色体状态
交叉操作需要生成一个随机数num_positions,这个随机数的位置介于基因总数的1/3~2/3区间的位置,这个随机数代表了有多少父染色体的基因要按位置遗传给子染色体。具体代码如下:
/* num_positions决定了父染色体遗传多少基因给子染色体 */
num_positions = geqo_randint(root, 2 * num_gene / 3, num_gene / 3);
/* 选择随机位置*/
for (i = 0; i < num_positions; i++)
{
/* 随机生成位置,将父染色体这个位置的基因遗传给子染色体 */
pos = geqo_randint(root, num_gene - 1, 0);
offspring[pos] = tour1[pos];
/* 标记这个基因已经使用,母染色体不能再遗传相同的基因给子染色体 */
city_table[(int) tour1[pos]].used = 1;
}
假设父染色体需要遗传2个基因给子染色体,分别传递第1号基因和第2号基因,那么子染色体目前的状态如图6-17所示。

图6-17 当前染色体状态
目前子染色体已经有了3和2两个基因,则母染色体排除这两个基因后,还剩下1和4两个基因,将这两个基因按照母染色体中的顺序写入子染色体,新的子染色体就生成了,如图6-18所示。

图6-18 新的染色体装状态
5. 适应度计算
/* 适应度分析*/
kid->worth = geqo_eval(root, kid->string, pool->string_length);
/* 基于适应度将染色体扩散*/
spread_chromo(root, kid, pool);
由于种群中的染色体始终应保持有序的状态,spread_chromo函数可以使用二分法遍历种群来比较种群中的染色体和新染色体的适应度大小并根据适应度大小来查找新染色体的插入位置,排在它后面的染色体自动退后一格,最后一个染色体被淘汰,如果新染色体的适应度最大,那么直接会被淘汰。具体代码如下:
/* 二分法遍历种群Pool中的染色体 */
top = 0;
mid = pool->size / 2;
bot = pool->size - 1;
index = -1;
/* 染色体筛选 */
while (index == -1) {
/* 下面4种情况需要进行移动*/
if (chromo->worth <= pool->data[top].worth) {
index = top;
} else if (chromo->worth - pool->data[mid].worth == 0) {
index = mid;
} else if (chromo->worth - pool->data[bot].worth == 0) {
index = bot;
} else if (bot - top <= 1) {
index = bot;
} else if (chromo->worth < pool->data[mid].worth) {
/*
* 下面这两种情况单独处理,因为他们的新位置还没有被找到
*/
bot = mid;
mid = top + ((bot - top) / 2);
} else { /* (chromo->worth > pool->data[mid].worth) */
top = mid;
mid = top + ((bot - top) / 2);
}
}
遗传算法会通过选择优秀的染色体以及多次的代际交叉,不断地生成新种群的染色体,循环往复,推动算法的解从局部最优向全局最优逼近。
6.4 小结
本章对SQL引擎的主要实现流程进行了介绍,包括SQL解析流程、查询重写、查询优化等。SQL引擎内容较多代码耦合度高,实现逻辑较为复杂,需要读者对代码整体流程以及关键结构体有所掌握,并在应用实践中不断总结才能做到更好的理解。
感谢大家学习第六章SQL引擎源解析的内容,下一篇我们开启第七章执行器解析的相关精彩内容介绍。敬请期待。
