




《扩增子16S/18S/ITS-QIIME2 详细实战手册》

发现“分享”和“赞”了吗,戳我看看吧
目录
前言(简介)
目前主流插件
常用语义类型
DADA2分析常用流程
1.环境配置、数据准备和和文件路径设置
2.数据分析开始(导入双端序列)
3.生成特征表和代表序列(DADA2)
4.物种分类与组成(过滤污染序列、叶绿体、线体体、古菌和稀有物种,构建进化树)
5.QIIME2多样性分析(不同水平分类汇总、物种组成差异分析、alpha和beta多样性分析、多样性稀释曲线)
6.导出unifrac距离矩阵文件
7.数据提取,方便后续个性化分析(导出特征表、注释信息、进化树和代表序列)
Cutadapt+Deblur分析常用流程
1.环境配置、数据准备和和文件路径设置
2.数据分析开始(cutadapt剪切引物信息、双端合并和质控过滤数据)
3.生成特征表和代表序列(方法1:导入其他软件分析好的table与代表序列。方法2:Deblur 操作命令质控,命令Deblur 16S用于16S序列和命令Deblur other完成ITS分析)
4.物种分类与组成(过滤污染序列、叶绿体、线体体、古菌和稀有物种,构建进化树)
5.QIIME2多样性分析(不同水平分类汇总、物种组成差异分析、alpha和beta多样性分析、多样性稀释曲线)
6.导出unifrac距离矩阵文件
7.数据提取,方便后续个性化分析(导出特征表、注释信息、进化树和代表序列)
“前言”
该流程可在Linux或Mac系统中运行,相比mothur具有更多的优点,主要包括:整合了200多款相关软件和包,实现每个步骤更多软件和方法的选择;提供150多个脚本,实现各种个性化分析,并可以应对不同类型数据和实验设计;流程开放程度高,容易整合新软件和方法;增强统计和可视化,实现多样性、物种组成、差异比较和网络等众多方法和出版级图表绘制。由于QIIME允许同领域研究者较自主地开展扩增子数据的个性化分析和可视化,逐渐成为本领域最受欢迎的软件。为了满足日益增长的测序数据量和可重复计算的要求,Gregory J. Caporaso教授于2016年起发起了基于Python3语言从头编写的QIIME2项目。该项目实现了分析流程的可追溯以满足科研可重复计算的要求;同时推出了一系列新算法,如基于进化距离的快速算法条型(Striped)UniFrac、物种分类新方法2-feature-classifier等;更重要的是软件的可扩展性和得到了同行的广泛支持,如接头和引物序列去除工具cutadapt、序列质量控制R包DADA2、聚类和去冗余的软件VSEARCH、纵向和成对样本分析工具longitudinal等,甚至包括宏基因组、宏代谢组分析和中文帮助文档,极大了提高了流程的适用范围和易用性。
QIIME2插件多种质量控制并生成特征表的方式主要有两种,一种是通过去噪,即生成扩增/绝对序列变体(Absolute Sequence Variants,ASV),ASV是最近发展的新一代方法,在功能上提供更好的分辨率。ASV可以基于400bp或更多序列中单个核苷酸的差异来分离特征,甚至超过99%同一性OTU聚类的分辨率。目前在QIIME 2 中可通过DADA2(q2-dada2)和Deblur(q2-deblur)插件实现。
简单地说,分为主流两个部分来说;
DADA2和Deblur;
注意:介绍内容大部分来自微信公众号“环微分析”,加上小编自己的分析流程,可以使故事说的更加圆满。
目前主流插件
alignment: 用于生成和处理序列对齐。
composition: 用于组合数据分析。
cutadapt: 用于从序列数据中删除适配器序列、引物和其他不需要的序列。
dada2: 使用dada2进行序列质量控制。
deblur: 使用deblur进行序列质量控制。
demux: 用于拆分序列和查看序列质量的插件。
diversity: 用于探索群落多样性。
diversity-lib: 用于计算群落多样性。
emperor: 用于排序绘图。
feature-classifier: 用于训练分类器。
feature-table: 用于按特征表处理样本。
fragment-insertion: 用于扩展系统发育。
gneiss: 用于构建成分模型。
longitudinal: 用于配对样本和时间序列分析。
metadata: 用于处理元数据。
phylogeny: 生成和处理系统发育。
quality-control: 用于特征和序列数据质量控制。
quality-filter: 用于基于PHRED的过滤和修整。
sample-classifier: 用于对样本元数据进行机器学习预测。
taxa: 用于处理功能分类注释的插件。
types: 用于微生物组分析的类型定义。
vsearch: 用于通过vsearch进行聚类和去冗余。
常用语义类型
FeatureTable[Frequency]:特征表[频率],即OTU表,其中每个值表示对应样本中OTU的出现频率。
FeatureTable[RelativeFrequency]: 特征表[相对频率],其中每个值表示相应样本中OTU的相对丰度,使得每个样本的值之和为1.0。
FeatureTable[PresenceAbsence]: 特征表[存在/缺席],其中每个值表示相应样本中是否存在某个OTU。
FeatureTable[Composition]: 特征表[组成],其中每个值表示相应样本中OTU的频率,并且所有频率都大于零。
Phylogeny[Rooted]: 系统发育[根],有根的系统发育树。
Phylogeny[Unrooted]: 系统发育[无根],无根的系统发育树。
DistanceMatrix: 距离矩阵。
PCoAResults: 主坐标分析PCoA的结果。
SampleData[AlphaDiversity]: 样本数据[Alpha多样性],每个数值均为Alpha多样性结果,基于样本自身的分析。
SampleData[SequencesWithQuality]: 样本数据[带质量的序列],要求序列有质量值,要求序列名称与样品存在对应关系,如为按样品拆分后的数据格式。
SampleData[PairedEndSequencesWithQuality]: 样本数据[带质量的成对末端序列],要求序列ID与样品编号存在对应关系。
FeatureData[Taxonomy]: 特征数据[分类学],每一个特征的分类学信息。
FeatureData[Sequence]: 特征数据[序列],代表性序列。
FeatureData[AlignedSequence]: 特征数据[对齐序列],代表性序列进行多序列比对的结果。
FeatureData[PairedEndSequence]: 特征数据[双端序列],双端序列进行聚类或去噪后,生成的OTU/Feature。
EMPSingleEndSequences: 采用地球微生物组计划标准实验方法产生的单端测序数据。
EMPPairedEndSequences: 采用地球微生物组计划标准实验方法产生的双端测序数据。
TaxonomicClassifier:一种经过训练的分类器,用于物种注释。
DADA2分析常用流程
……
DADA2质量控制过程将过滤掉在测序数据中鉴定的任何phiX序列(最主要的目的:
1.调节碱基平衡,改善测序仪的空间校正,便于后期提高base calling的准确性;
2.由于Phix序列已知基因组较小,在测序的过程中Illumina的测序仪就开始将测的read与phix基因组进行比较,预估测序指标。)通常存在于标记基因Illumina测序数据中,用于提高扩增子测序质量,并同时过滤嵌合序列(即嵌合基因,就是两个基因共用一段DNA序列,这两个基因称为嵌合基因)。
在DADA2中,双端合并,去除嵌合体,截去接头序列降噪生成feature table都是一步完成的。所以,运行DADA2之前要确保测序数据满足以下规范:
①样品已被拆分好,即每个样品一个fq/fastq文件(或者双端成对fq文件);
②已经去除非生物核酸序列,比如:引物(primers),接头(adapters or barcodes),linker等;
③如果样品是下机的双端测序,其应具有双端测序的相匹配的两个fq文件。
#1.环境配置、数据准备和导入
备注:
小编这里QIIME2分析放在文件夹[All]-Qiime2中,[All]-Qiime2/创建自己分析目录,这里以2022.9.18文件夹为例,后续读者可自行修改。
其他分析数据可以按照小编这样顺序放置。
然后再在分析目录中,创建0.raw_data存放原始数据,注意的是,文件R1和R2名字必须与manifest文件中对应起来,否则出现QIIME2导入不进去数据。
在QIIME 2中,所有数据都被构造为特定语义类型的对象。使用样本清单格式(manifest format)导入序列,这是一种在QIIME 2中导入拆分样本数据的通用方法。普通用户常用的下机数据格式为.fastq文件,需要创建一个清单文件,然后使用qiime tools import命令手动输入。清单文件是一个文本文件(.tsv或.txt格式),它将示例标识符映射到fastq.gz或fastq的绝对文件路径,其中包含示例的序列和质量数据。清单文件还指示每个fastq.gz或fastq文件中的读取方向。
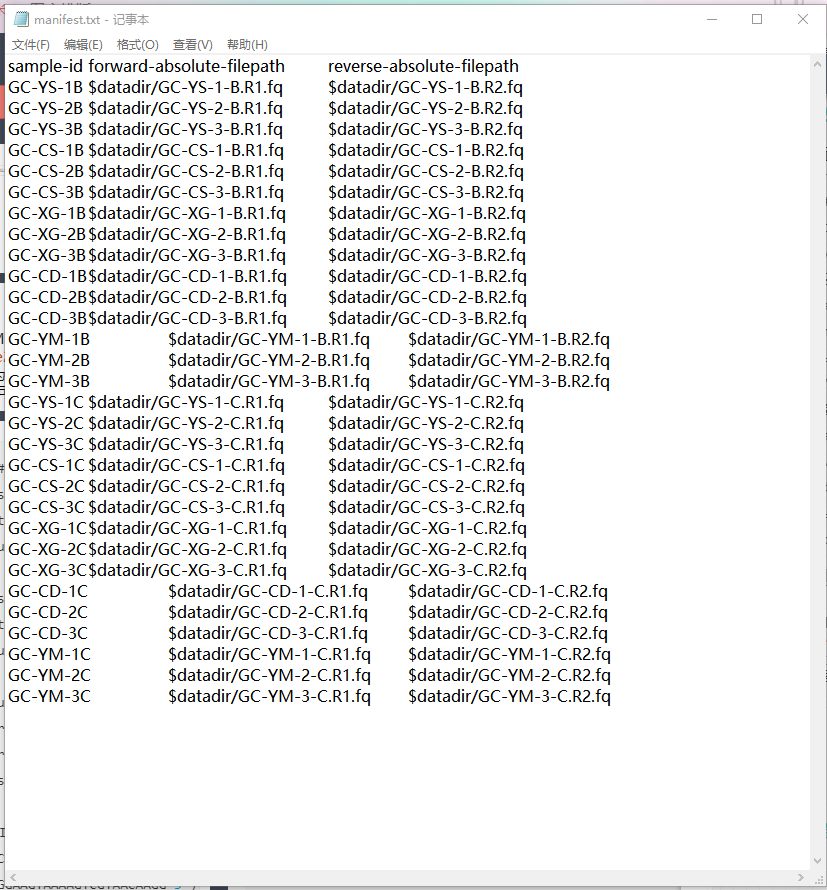
原始序列位置信息manifest.txt,保存格式为UTF-8,另外,采用制表符Tab将信息隔开,在txt打开如图下所示,可能肉眼觉得有些信息没有隔开。
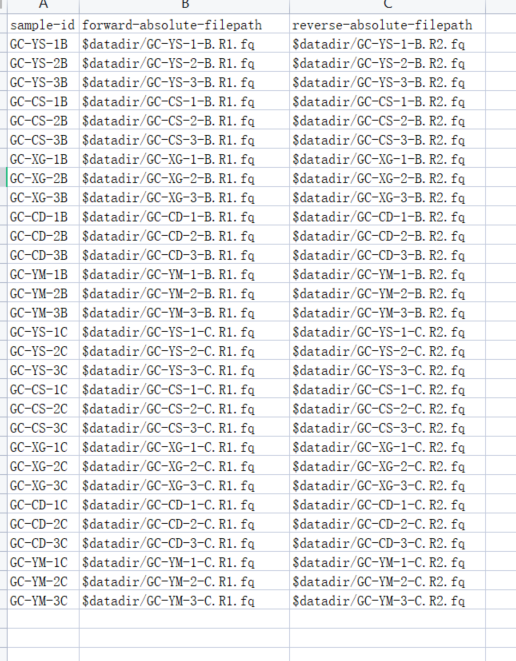
但是,在wps打开,如图下所示
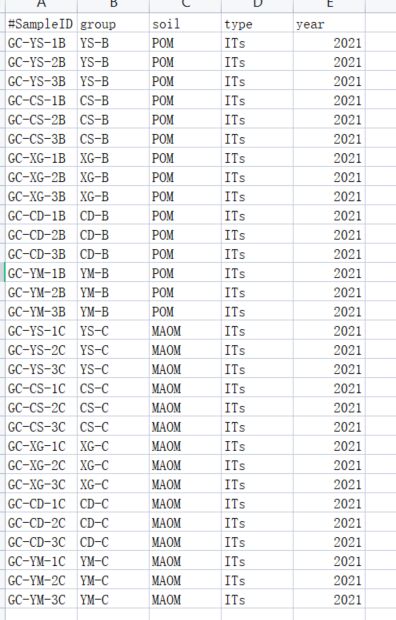
分组信息表fastmap.txt保存格式为UTF-8,另外,采用制表符Tab将信息隔开。

#文件路径
#####################################################启动qiime2环境中conda activate qiime2-2021.2#设置工作目录及名字working__name=DADA2-16Sworkdir=/mnt/e/[All]-Qiime2/$working__name#数据库所在的路径dbdir=/mnt/e/[All]-Qiime2/qiime2_database/tlc_classifier#这里指定测序数据所在的路径,并export为全局变量,manifest文件中才好找到数据文件($datadir)export datadir=/mnt/e/[All]-Qiime2/$working__name/0.raw_datamanifest=$workdir/manifest.txtecho $datadir#实验设计文件路径fastmap=$workdir/fastmap.txt
#2.数据分析开始
##########################################################数据分析开始###########################################################cd $workdirmkdir $workdir/1.import_datacd $workdir/1.import_data## 1. 导入双端测序数据##导入数据;time qiime tools import \--type 'SampleData[PairedEndSequencesWithQuality]' \--input-path $manifest \--output-path paired-end-demux.qza \--input-format PairedEndFastqManifestPhred33V2#数据质量查看;qiime demux summarize \--i-data paired-end-demux.qza \--o-visualization paired-end-demux.qzv
使用qiime demux summarize命令检查样本的序列和测序深度(它提供每个样本中序列数及序列质量的信息)
#3.生成特征表和代表序列(DADA2)
命令注释:
①在使用qiime dada2 denoise-single/ qiime dada2 denoise-paired时可设置--p-n-threads 参数,用于设置运行时使用的线程数量。线程越多,则运行速度越快。当线程设置为0时则默认使用全部线程;
②--p-trim-left截取左端低质量序列,有时用于切除低质量序列、barocde或引物。查看demux_seq.qzv文件中的箱线图,左端质量都很高,无低质量区,设置为0;或可直接忽略此参数设置;
③--p-trunc-len序列截取长度,也是为了去除右端低质量序列,我们看到大于150以后,质量下降极大,甚至中位数都下降至20以下,需要全部去除,综合考虑决定设置为150;
④当处理双端数据时,需考虑截取后的序列是否可以成功拼接。目前最短的拼接长度为引物长度+12bp。
## 2. 生成特征表和代表序列cd $workdirmkdir $workdir/2.feature_table/cd $workdir/2.feature_table/###dada2数据质量过滤,合并,去嵌合,去引物一步完成;qiime dada2 denoise-paired \--i-demultiplexed-seqs $workdir/1.import_data/paired-end-demux.qza \--p-n-threads 16 \--p-trim-left-f 29 --p-trim-left-r 18 \--p-trunc-len-f 0 --p-trunc-len-r 0 \--o-table dada2-table.qza \--o-representative-sequences dada2-rep-seqs.qza \--o-denoising-stats dada2-stats.qza###分析结果可视化qiime metadata tabulate \--m-input-file dada2-stats.qza \--o-visualization dada2-stats.qzvqiime feature-table summarize \--i-table dada2-table.qza \--o-visualization dada2-table.qzv
#4. 物种分类与组成
这里注释文件见教程2-QIIME2 物种注释数量训练集
## 3. 物种分类与组成cd $workdirmkdir $workdir/3.asv_taxonomy/cd $workdir/3.asv_taxonomy/### 物种注释 16S#特异引物作为分类数据库###细菌注释(可选1)特定引物的分类数据库(V4-V5):qiime feature-classifier classify-sklearn \--i-classifier $dbdir/classifier_gg_13_8_99_V4-V5.qza \--i-reads $workdir/2.feature_table/dada2-rep-seqs.qza \--p-n-jobs 16 \--o-classification taxonomy.qza###细菌注释(可选2) 物种注释16S#全长作为分类数据库gg,用Sliva全长容易报错;qiime feature-classifier classify-sklearn \--i-classifier $dbdir/classifier_gg_13_8_99.qza \--i-reads $workdir/2.feature_table/dada2-rep-seqs.qza \--p-n-jobs 16 \--o-classification taxonomy.qza##可视化物种注释qiime metadata tabulate \--m-input-file taxonomy.qza \--o-visualization taxonomy.qzv###对数据进行过滤#16s分析 过滤污染 叶绿体 线粒体 古菌qiime taxa filter-table \--i-table $workdir/2.feature_table/dada2-table.qza \--i-taxonomy taxonomy.qza \--p-exclude mitochondria,chloroplast,Archaea \--p-include p__ \ #过滤p_未注释的信息(备选)--o-filtered-table dada2-table-filt-contam.qza#ITs分析 过滤污染 叶绿体 线粒体qiime taxa filter-table \--i-table $workdir/2.feature_table/dada2-table.qza \--i-taxonomy taxonomy.qza \--p-exclude mitochondria,chloroplast \--p-include p__ \ #过滤p_未注释的信息(备选)--o-filtered-table dada2-table-filt-contam.qza#过滤稀有ASV,可查看dada2-table.qzv 确定过滤数量:--p-min-frequency为所有tags数的0.00005qiime feature-table filter-features \--i-table dada2-table-filt-contam.qza \--p-min-frequency 50 \ #总序列数*万分之一--p-min-samples 1 \--o-filtered-table dada2-table-final.qza#更新一下repset-seqs.qza,将过滤掉的ASV对应的序列过滤掉qiime feature-table filter-seqs \--i-table dada2-table-final.qza \--i-data $workdir/2.feature_table/dada2-rep-seqs.qza \--o-filtered-data dada2-repset-seqs-final.qza##构建进化树16S分析,ITS一般不构建进化树qiime phylogeny align-to-tree-mafft-fasttree \--i-sequences $workdir/3.asv_taxonomy/dada2-repset-seqs-final.qza \--o-alignment aligned-rep-seqs.qza \--p-n-threads 16 \--o-masked-alignment masked-aligned-rep-seqs.qza \--o-tree unrooted-tree.qza \--o-rooted-tree rooted-tree.qza##过程中.qza和.qzv文件都可以放到view.qiime2.org这个网站上生成图### 查看特征表信息qiime feature-table summarize --i-table $workdir/3.asv_taxonomy/dada2-table-final.qza \--o-visualization dada2_table_final_summary.qzv \--m-sample-metadata-file $fastmap
#5.qiime2分析多样性
##4.qiime2分析多样性cd $workdirmkdir $workdir/4.qiime2_diversity_analysiscd $workdir/4.qiime2_diversity_analysis##物种组成分析qiime taxa barplot \--i-table $workdir/3.asv_taxonomy/dada2-table-final.qza \--i-taxonomy $workdir/3.asv_taxonomy/taxonomy.qza \--m-metadata-file $fastmap \--o-visualization taxa-bar-plots.qzv####不同分类水平分类汇总qiime taxa collapse \--i-table $workdir/3.asv_taxonomy/dada2-table-final.qza \--i-taxonomy $workdir/3.asv_taxonomy/taxonomy.qza \--p-level 6 \--o-collapsed-table feature-table-final-L6.qza##物种组成差异分析:ANCOM方法假定数据中差异的物种少于25%,如果你觉得自己的数据差异很大就不能使用ANCOM;###由于composition不能处理为0的数据,转换+1#格式化特征表,添加伪计数qiime composition add-pseudocount \--i-table $workdir/3.asv_taxonomy/dada2-table-final.qza \--o-composition-table comp-table.qza###差异比较(注意group分组信息,表头要对)qiime composition ancom \--i-table comp-table.qza \--m-metadata-file $fastmap \--m-metadata-column group \--o-visualization ancom-type.qzv##############16s细菌分析#############################(细菌运行这步就行)###alpha和beta多样性分析;这个49125根据dada2-table.qzv中的Minimum frequency值qiime diversity core-metrics-phylogenetic \--i-phylogeny $workdir/3.asv_taxonomy/rooted-tree.qza \--i-table $workdir/2.feature_table/dada2-table.qza \--p-sampling-depth 49125 \--m-metadata-file $fastmap \--output-dir core-metrics-phylogenetic###多样性稀释曲线;这个127291根据dada2-table.qzv中的Maximum frequency(通用)qiime diversity alpha-rarefaction \--i-table $workdir/2.feature_table/dada2-table.qza \--i-phylogeny $workdir/3.asv_taxonomy/rooted-tree.qza \--p-max-depth 127291 \--m-metadata-file $fastmap \--o-visualization alpha-rarefaction.qzv##差异比较###alpha多样性比较qiime diversity alpha-group-significance \--i-alpha-diversity $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/observed_features_vector.qza \--m-metadata-file $fastmap \--o-visualization observed_features-group-significance.qzv###beta多样性比较qiime diversity beta-group-significance \--i-distance-matrix $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/bray_curtis_distance_matrix.qza \--m-metadata-file $fastmap \--m-metadata-column Group \--o-visualization bray_curtis-Group-significance.qzv \--p-pairwise
#6.导出unifrac距离矩阵文件
##5.导出unifrac距离矩阵文件cd $workdirmkdir $workdir/5.unifrac_analysiscd $workdir/5.unifrac_analysis#导出无权重unifrac距离矩阵qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/unweighted_unifrac_distance_matrix.qza \--output-path unweighted_unifrac_distance_matrix#导出有权重的unifrac距离矩阵qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/weighted_unifrac_distance_matrix.qza \--output-path weighted_unifrac_distance_matrix#导出jaccard距离矩阵qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/jaccard_distance_matrix.qza \--output-path jaccard_distance_matrix.qza#导出Bray-Curtis距离矩阵qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/bray_curtis_distance_matrix.qza \--output-path bray_curtis_distance_matrix#导出香浓指数qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/shannon_vector.qza \--output-path shannon_vector#导出Alpha多样性考虑进化的faith指数qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/faith_pd_vector.qza \--output-path faith_pd_vector#导出Alpha多样性observed otus指数qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/observed_features_vector.qza \--output-path observed_features__vector#导出Alpha多样性均匀度指数qiime tools export \--input-path $workdir/4.qiime2_diversity_analysis/core-metrics-phylogenetic/evenness_vector.qza \--output-path evenness_vector
#7.数据提取,方便后续个性化分析
###qiime2特有的qza,qzv文件其实是zip压缩文件,可以将qza或者qzv后缀名改成zip,解压即可查看cd $workdirmkdir $workdir/6.export_datacd $workdir/6.export_data#导出特征表为biom格式qiime tools export \--input-path $workdir/3.asv_taxonomy/dada2-table-final.qza \--output-path $workdir/6.export_databiom convert -i feature-table/dada2-table.biom \-o dada2-table.txt \--to-tsv#删除注释行sed -i '/# Const/d' eature-table.txt##导出特征表***重要文件***便于后续分析##导出注释信息qiime tools export \--input-path $workdir/3.asv_taxonomy/repset-seqs-final.qza \--output-path $workdir/6.export_data##导出进化树(真菌不做进化树,因为保守区)qiime tools export \--input-path $workdir/3.asv_taxonomy/rooted-tree.qza \--output-path $workdir/6.export_data##导出代表序列qiime tools export \--input-path $workdir/3.asv_taxonomy/dada2-repset-seqs-final.qza \--output-path $workdir/6.export_data# 完

扫二维码,关注我们
微信号|KSM生信分析
加油,陌生人!
文案撰写:玖柒年
排版编辑:玖柒年
封面审核:玖柒年
