




公司内部的一个技术分享,感谢培训小姐姐同意可以让我po到自己的公众号上。
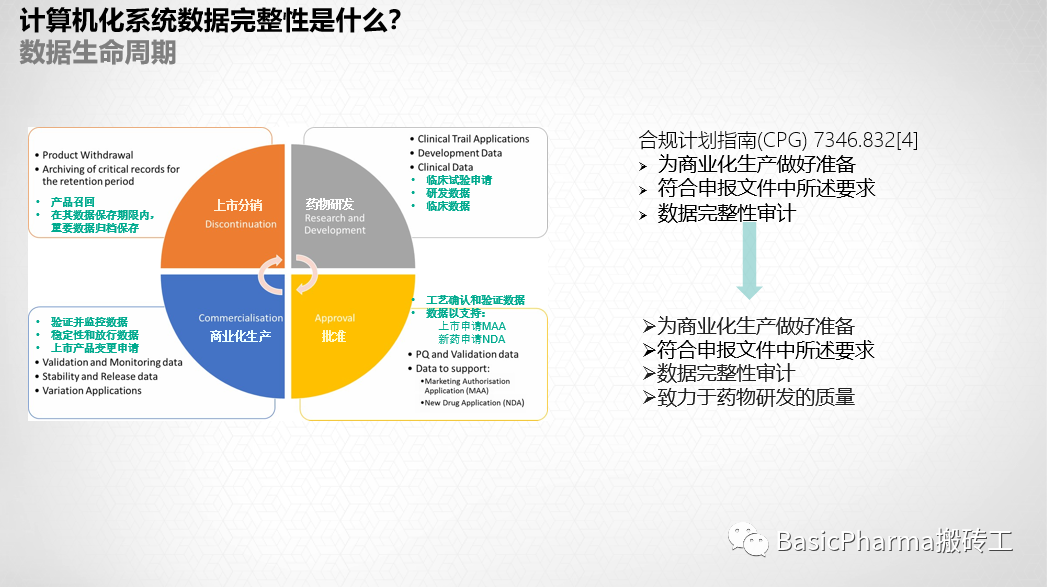
数据治理和完整性将应用于整个产品生命周期()。它始于药品研发阶段,并持续到研发的早期和后期阶段。完整的GMP和DI要求将在产品分销和商业化时应用。针对数据完整性的数据治理需要在整个生命周期阶段一致地应用,并根据gmp要求的范围进行应用。需要根据药品质量体系(PQS)的要求,在药品生命周期的不同阶段,定义并系统应用基于风险的方法的全面模型。特别是,与临床、药品研发和商业化的产品/流程相比,药品研发部门对数据完整性的要求不同且不那么严格。DI的整体模型需要确保相关数据从研发和商业生命周期的所有阶段都是可用的,直到产品超过追溯期,数据可销毁为止。
由于数据造假或数据管理实践不良,GMP监管实验室的数据完整性是当前监管机构的热门话题。数据完整性不仅仅局限于一个国家或大陆,而是一个全球问题,因为许多数据完整性问题是基于糟糕和/或过时的工作实践,而不是少数涉及数据伪造的案例。
实验室的数据完整性可以追溯到20世纪90年代初的巴尔实验室。在这里,生产中的一个问题被追踪到质量控制实验室,在那里发现实验室正在重新测试和重采样,直到批次通过。在法院判决之后,法官裁定除非得到美国药典(USP)的允许,否则异常值不能被拒绝[1]。FDA也在1993年发布了《药品质量控制实验室检查指南》[2]。本指南仍然是相关的,因为在受监管的实验室中,许多过程仍然是基于纸质的或使用混合系统,应该阅读本文件,因为它提供了有价值的见解,监管机构将如何进行质量控制实验室的检查。
2005年,Able Laboratories欺诈案是一个重大事件,该案件发现了操纵或伪造数据以通过[3]。美国食品和药物管理局(FDA)最担心的是,问题不是由他们的检查员发现的,而是由公司的举报人发现的。对FDA来说不幸的是,Able实验室有7次成功的预批准检查(PAI)。直接的结果是,FDA完全重写了批准前检查的合规计划指南(CPG) 7346.832[4],以便能够在PAI期间发现类似的问题。更新后的CPG于2012年5月生效。这版本的CPG有三个目标:
1.Readiness for Commercial Manufacturing
为商业化生产做好准备
1.Conformance to the Application
符合申报文件中所述要求
1.Data Integrity Audit
数据完整性审计
乍一看,实验室数据完整性的重点是目标3。然而仔细阅读CPG[4],人们会意识到实验室和生产数据完整性渗透到所有三个目标中,仅关注实验室数据的目标3是不够的。(西门:此文件已更新)
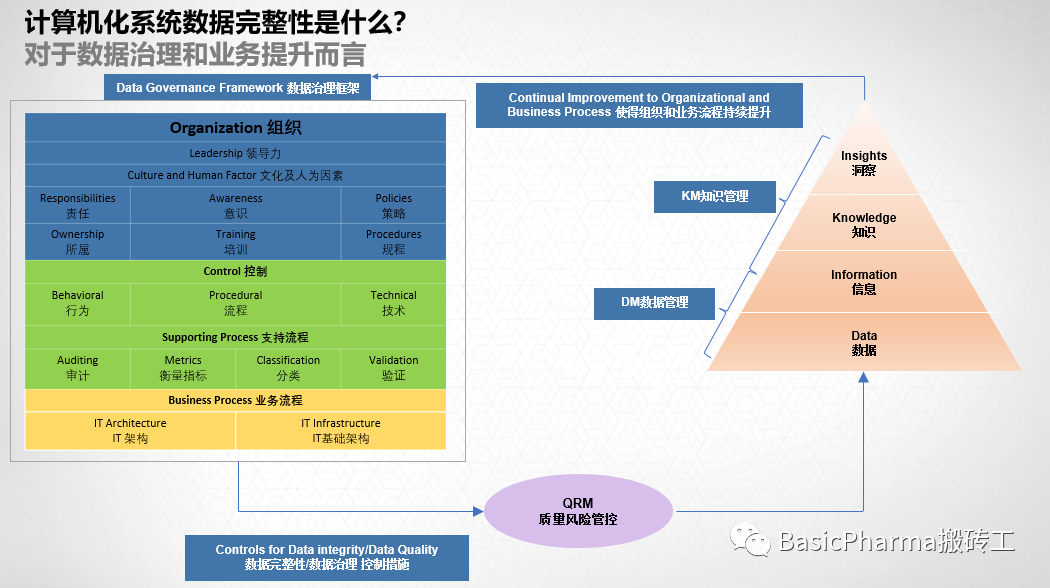
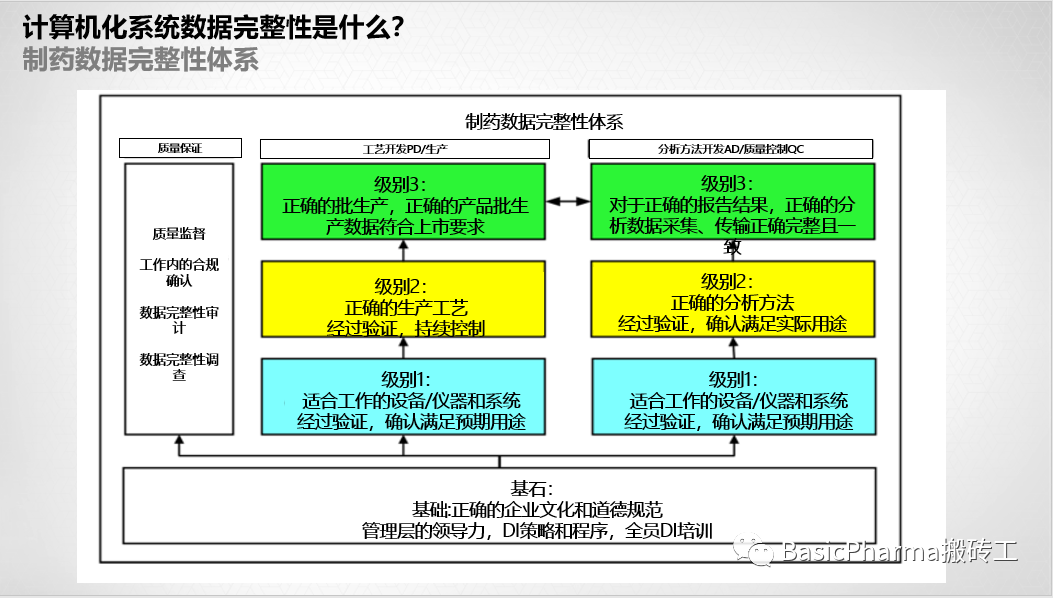
数据完整性必须理解在药品质量体系支持下,经过验证和确认的分析过程中,从经过验证的生产操作中提取的代表性样品的分析背景下[8,9]。数据完整性并不存在于真空中。
基础:正确的企业文化和道德规范
这个数据完整性模型的基础是组织中所有人(含管理层)共同参与的。这是为了确保数据完整性/数据治理在药品质量体系的背景下得到牢固的确立。因此,必须有管理领导,企业数据完整性政策,关联到实验室和生产数据完整性程序,员工进行初始和持续的数据完整性培训。
组织内所有员工的共同参与对确保数据完整性是至关重要的。FDA在PQS指南和EU GMP第1章[8,9]中明确指出,管理层对组织内部的质量负责,其中包括数据完整性。
级别1:适合工作的设备/仪器和系统
如果所使用的分析仪器不充分合格,或者控制仪器或处理数据的软件没有经过验证,那么进行任何分析都是没有意义的。与过程和系统的互连需要被考虑在内(图4)。对于生产设备和控制软件系统也可以做出类似的评论。因此,在级别1中,用于药品生产和质量控制的分析仪器、生产设备和计算机化系统必须分别符合规定的操作范围,并被确认为“符合用户预期用途”。
第2级:正确的生产工艺和分析程序
在完成分析仪器和验证软件的确认后,即可以展开分析程序的开发并进行验证。ICH Q2(R1)[13]和欧洲药典(EP)和美国药典(USP)的相关章节对此有一些已发表的。然而,这些出版物的重点是验证已经开发的分析程序。
方法开发,这是非常重要的,因为它决定了整个过程的健壮性robust或坚固性,在这些出版物中很少受到注意。然而,随着Martin等人[14]在2012年发表了对USP通则关于这个主题的修订建议,这个分析领域正在发生变化。
目前正在起草一个药典通则,重点关注验证最佳实践,将于2018年发布。这意味着定义过程设计空间的“好的”科学合理的方法开发现在变得重要,因为在设计空间内对已验证方法的更改将被视为已验证本身。(西门君:USP1220 2022版)
级别3:正确批次的正确生产-正确报告结果的正确分析。
最后,在数据完整性模型的级别3,将执行实际的药品工作:批生产或分析样本。样本的分析必须使用正确的方法和正确的数据系统,由工作人员在一个能够以安全、准确、易读、同步、原始和可归因的方式生成、解释数据和计算可报告结果的环境中进行。应该鼓励员工承认任何错误,必须建立一种不责怪的文化。同样重要的是不要忘记整体质量管理体系的重要性。
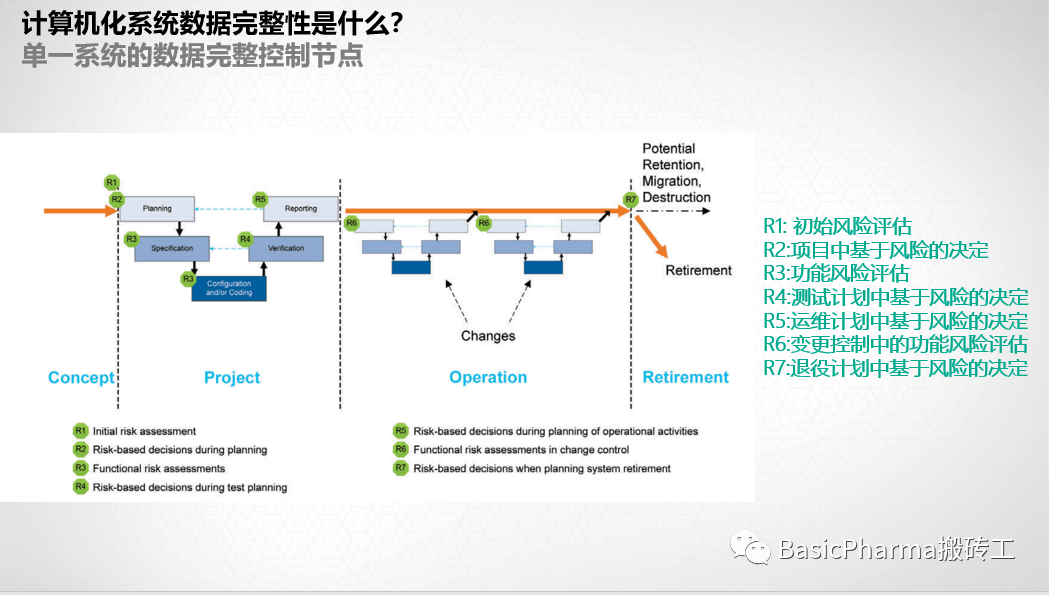
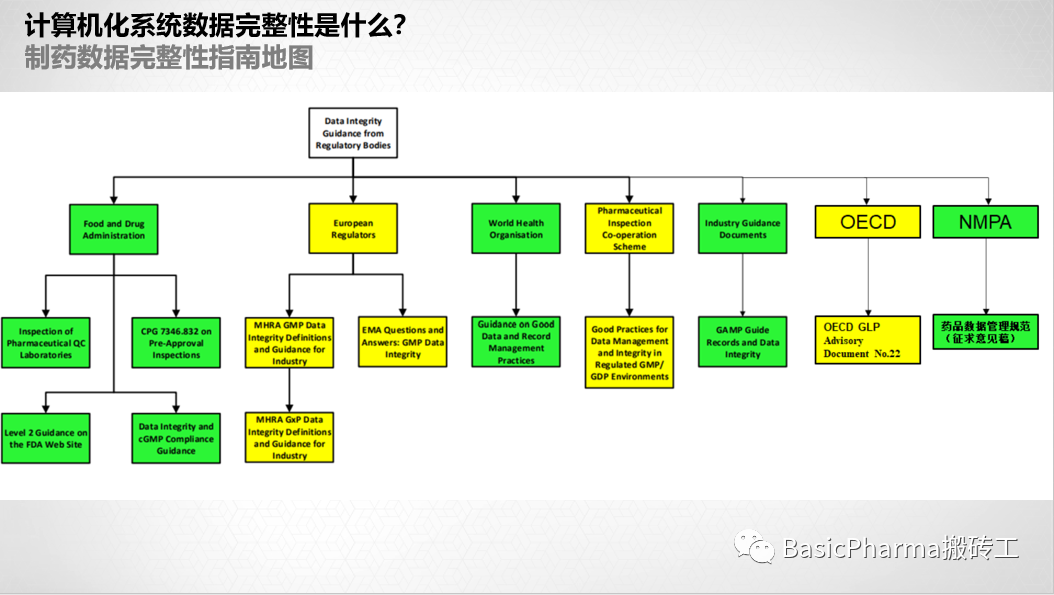
绿色仅针对GMP,黄色包含GLP

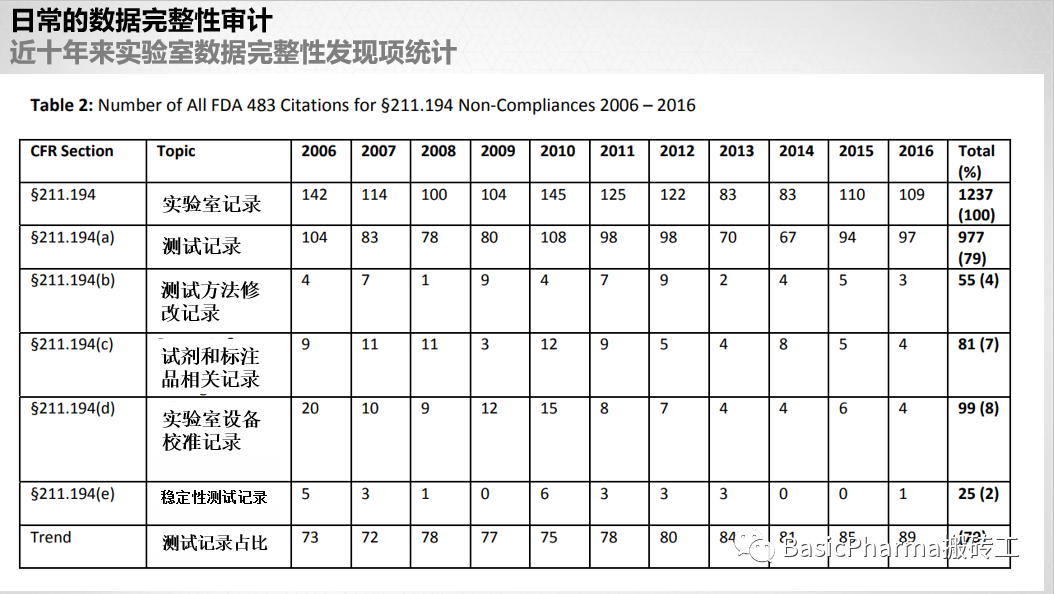
在过去的11年里,共有1237次对§211.194条款的引用,其中任何一年不符合的引用数量在83到145之间。
·然而,每个条款不遵守情况的分布更有趣。问题最多的明确区域是§211.194(a)测试记录,它构成了80%的不符合。
·在解释表格中的数据时,我们需要考虑到,引用的混合可能并不能完全反映小公司和大公司的市场构成。
对§211.194(A)各子条款内的数据进行进一步分析表明,这一领域监管性引用的三个主要原因是:
·未能充分识别所使用的测试方法
·未能记录分析过程中取样的重量
·没有审阅人签名

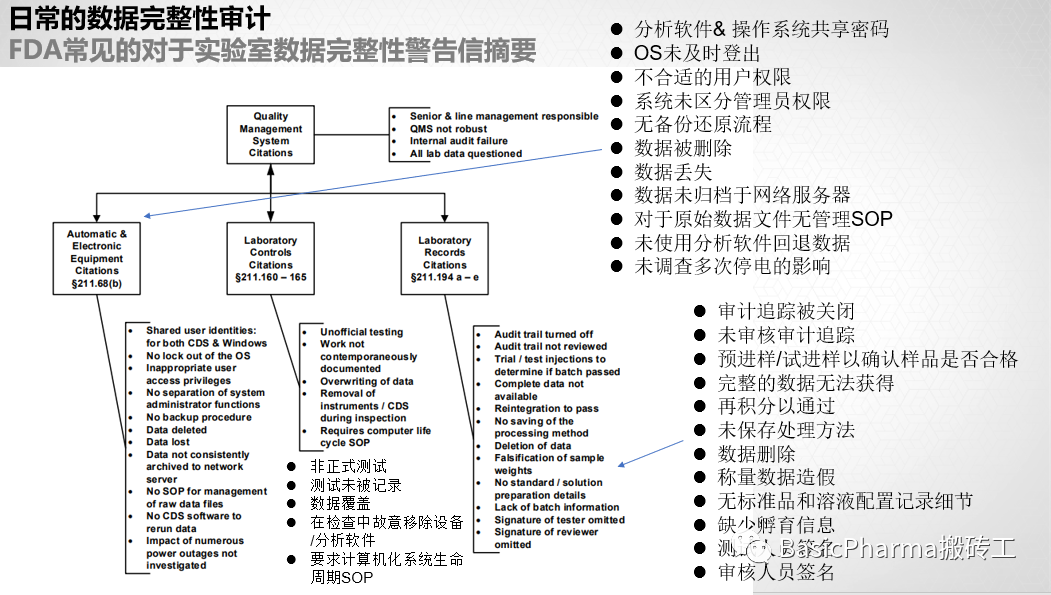
完整的数据和原始数据本质上是相同的术语,对于计算机化系统,包括审计追踪条目的复核,无论该系统是用作混合应用程序还是电子应用程序。如果应用程序支持这种方法,第二人复核的审计追踪的重点应该是例外审核。例外审核是一种基于风险的数据完整性方法,它只审查系统通知审查员的与gmp相关的修改或删除的适用审计追踪条目。这种工作方式可以是用颜色编码数据、复选框或注释等。
·在用户需求规范中指定功能
·记录应用程序配置
·验证功能在用户接收测试(性能确认)阶段是否正常工作
·通过例行的数据完整性审计和/或定期审查,确保配置设置未被更改
检查范围:
•数据缺失或不一致
•异常值数量过多(表示数据处理效率低下)
•测试结果的意外低可变性
•与协议或数据收集和报告的偏差过多
•中止运行次数过多
•测试顺序和时间安排:人们产生测试结果的速度是否比测试顺序允许的速度快?
•具有相同创建日期的重复文件
•分析师绩效:分析师绩效是否太好而无法实现(执行测试的时间/样本数量)?
•数据审查:审查过程是否太短?(表示数据审查中采用的快捷方式)
•上次访问系统与上次报告的测试结果(表明用户帐户已被劫持或共享给其他用户)
•过度的用户访问和角色更改:同一个用户ID是否来回更改多次?(表示利益冲突和可能操纵审计线索和文件)
•在CDS的情况下
o手动集成(集成直到通过行为),
o重新处理或修改的运行与正确的第一次运行(表明由于错误而需要重新处理的过程或样品的效率低下)
o短时间运行(可指示使用测试路径和测试是否符合要求)
o具有相同样本名称/批号的多个文件/运行(可指示第二次测试并测试是否符合要求)


